Research
Our research aims are mainly focused on the design, synthesis and application of molecular containers. We work in the synthesis of calix[4]pyrrole structures soluble in both organic and aqueous media. We study their binding processes with biologically relevant molecules (e.g. anions, N-oxides). This constitutes the starting point for further understanding more complex biological molecular recognition processes or the mechanism operating in sensing devices. We pursue the preparation of molecular capsules by using different strategies, including covalent chemistry, dynamic covalent bonds and lately metal-coordination bonds. The preparation of mechanically interlocked structures is another area of our interest. Rotaxanes and catenanes present unique three-dimensional cavities for anion recognition that resemble the preorganized pocket of anion binding proteins in Nature. That’s why we dedicate part of our efforts to synthesize and study the binding abilities of interlocked structures based on calix[4]pyrrole scaffolds. Finally, we also are proactive in the collaboration with other research groups working in the area of sensing devices. We want to apply the receptors prepared in the group for the development of sensing devices that can be used for the detection and quantification of clinically relevant molecules in real biological fluids (e.g. creatinine).
Synthesis of calix[4]pyrrole scaffolds
Our group has been interested in the synthesis of calix[4]pyrrole receptors for many years. The unique properties of the tetra-alpha isomer of aryl-extended calix[4]pyrroles have been exploited in several applications such as the self-assembly of molecular dimeric capsules, the transport of ions and ion-pairs across lipophilic membranes, and the development of new functional materials.
Aryl-extended calix[4]pyrroles are typically synthesized by the acid-mediated condensation reaction of an aryl methyl ketone with pyrrole. Generally, this reaction produces a mixture of the four different configurational calix[4]pyrrole isomers and a plethora of open oligomers. Most reported syntheses of arylextended calix[4]pyrroles either as pure isomers or isomeric mixtures target compounds with a simple methyl group attached to the mesocarbons. To date and to the best of our knowledge, only two examples featuring four substituents other than methyl groups have been reported. These examples described arylextended calix[4]pyrroles with a single type of functional group, either on the upper rim or the lower rim substituents. The condensation reaction of aryl alkyl ketones with pyrrole to produce the corresponding calix[4]pyrroles is significantly less efficient than the reaction with their aryl methyl counterparts, which may explain the limited examples. We designed an approach based on the simple addition of methyltrialkylammonium chloride salt to the acid-mediated condensation reaction of the aryl alkyl ketone and pyrrole. This methodology is by no means general, but it does provide modest to good yields of calix[4]pyrroles mainly possessing hydroxyl groups at their upper rims and terminal chloro or ester functions on their meso-alkyl substituents.

Water soluble calix[4]pyrroles
In recent years, the group has been interested in the synthesis of water soluble receptors and in the understanding of molecular recognition processes in aqueous media. We consider that the knowledge gained from the binding studies of simple water-soluble receptors could help in further understanding the operation of more complex biological systems.
In 2009, we reported the first example of a water-soluble aryl-extended calix[4]pyrrole bearing the water solubilizing groups at the upper rim. This specific location of the ionisable groups had several drawbacks: 1) alters the electronic nature of the pristine aromatic cavity and 2) impedes the further elaboration of the aromatic cavity with other functional groups capable of interacting with the included guest.
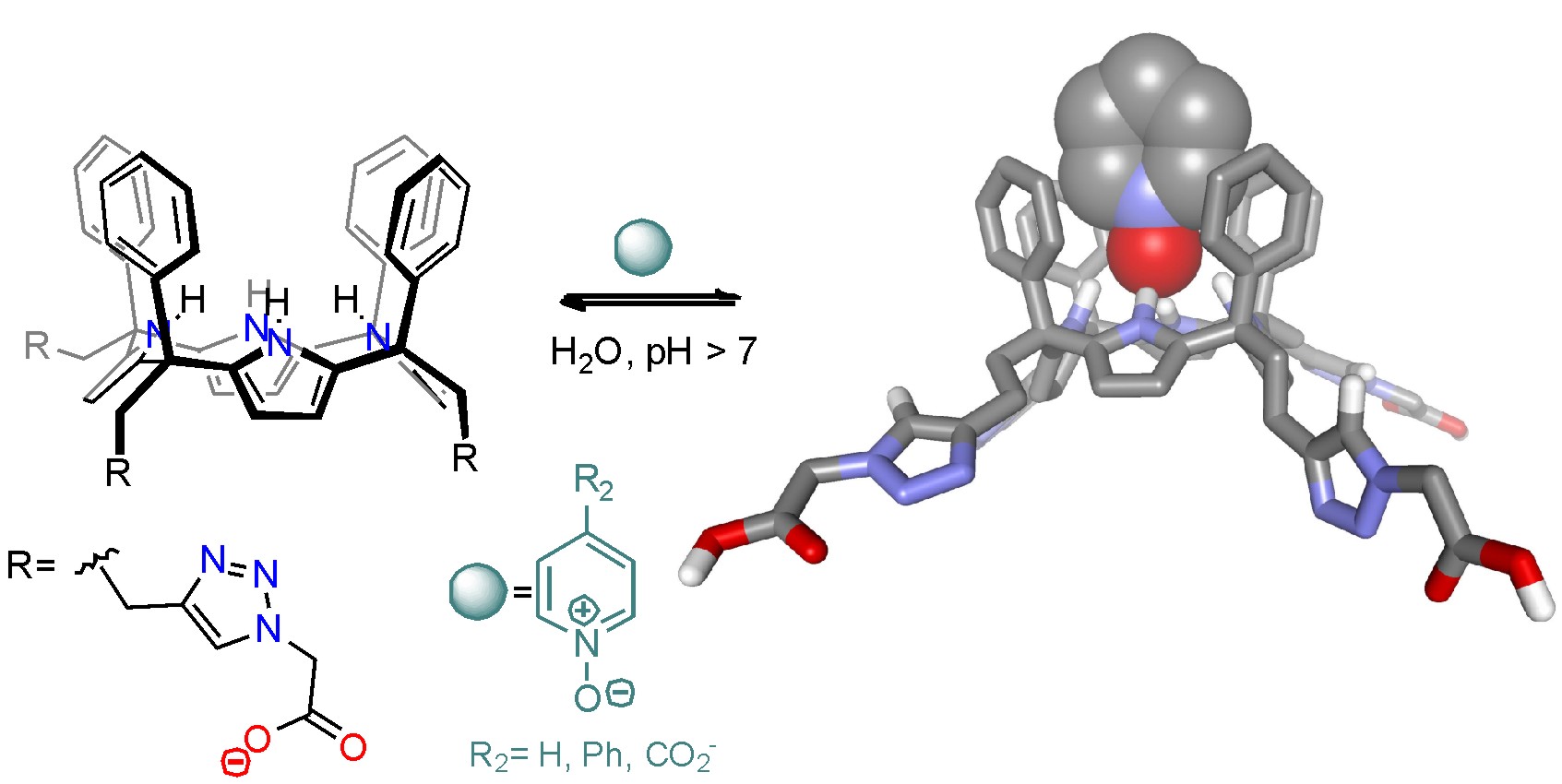
With the aim to overcome these limitations, the group described the synthesis of a water-soluble aryl-extended calix[4]pyrrole receptor bearing the water solubilizing groups at the lower rim. The solubilising groups were installed at the meso-alkyl substituents via 1,3-Huisgen cycloaddition. They are sufficiently distant to the binding site to avoid any interference in the recognition process. The receptor forms thermodynamically and kinetically stable complexes with a series of neutral and charged pyridyl N-oxide guests in basic aqueous solution with stability constants higher than 104 M-1. In all the cases, the N-oxide moiety engages in hydrogen-bond interactions with the four pyrrole NH’s of the calix[4]pyrrole core. Remarkably, the four-fold negatively charged water-soluble receptor is also able to bind an anionic pyridyl N-oxide guest equipped with a p-substituted carboxylate group with similar binding constant (>104 M-1).
These results demonstrate the ability of the receptor’s walls to protect the polar binding site and ensure the establishment of selective hydrogen-bonding interactions even in water solution.
Moreover, this year, we have reported the enlargement of the aromatic cavity defined by meso-phenyl substituents in the tetra-a isomers of aryl-extended calix[4]pyrroles affording unprecedented calix[4]pyrroles that we termed “super-aryl-extended” (SAE) (Fig. 1). We reported the binding properties of the prepared SAE-calix[4]pyrrole tetraester towards pyridyl-N-oxides.
The binding data revealed the formation of thermodynamically and kinetically highly stable 1:1 complexes. The complexation-induced chemical shifts indicated the formation of hydrogen bonds and aromatic interactions with the calix-core adopting the cone conformation. We quantified the additional interactions established between the four terminal aryl groups and the para-phenyl substituent of 4-phenyl pyridine N-oxide to be in the order of 1 kcalmol-1. Finally, to explore the water solubility of the SAE-calix[4]pyrroles, we appended ionizable groups at their upper rim. Unfortunately, we learnt that four ionizable groups was not enough to gain access to water solubility.

Dynamic covalent capsules
Molecular encapsulation has emerged as an attractive tool in host–guest chemistry to stabilize reactive intermediates, promote or accelerate chemical transformations and even alter their typical regio- and stereo-chemical outcomes. During the last decade, dynamic covalent bonds have been applied for thermodynamically controlled self-assembly processes of capsules and cages. This strategy combines the strength of covalent bonds with the reversibility and selectivity of non-covalent interactions. However, all the reported examples based on dynamic covalent bonds lack polar functions in their inner cavities. We have prepared polyimine molecular capsules based on calix[4]pyrrole scaffolds featuring large polar interiors. In brief, an aryl-extended tetraaldehyde calix[4]pyrrole scaffold was condensed with suitable diamines as linkers using templates (i.e. bispyridyl-N-oxides) for efficient self-assembly. The capsular complexes were characterized in solution, gas phase and the solid-state. Unprecedented transfer of asymmetry was observed from a chiral diamine linker to the resulting supramolecular capsular assembly. Current efforts are directed towards the reduction of the imine bonds to afford fully covalent capsules and the preparation of analogous water-soluble dynamic containers.

Siamese-twin calix[4]pyrrole tetramer
With the aim to extend the range of polar molecular containers based on aryl-extended calix[4]pyrrole scaffolds, we wanted to undertook the synthesis of an oligomacrocyclic calix[4]pyrrole dimer container using tetraalkynyl aryl-extended calix[4]pyrrole derivative as starting material. We expected that the incorporation of two additional aromatic walls to molecular container, compared to a previous one described in the group, would increase the thermodynamic and kinetic stability of its encapsulation complexes providing, simultaneously, a complete isolation of the bound guests from the bulk solution. The closed and polar internal cavity featured in the container suggested that it may have potential applications as a reactor vessel.
We demonstrated the importance that template molecules play in this reaction. After several attempts, we isolated an unexpected encapsulation complex of two bis-N-oxide guests in a tetrameric oligomacrocyclic calix[4]pyrrole structure.The obtained container is an unprecedented calix[4]pyrrole tetramer, in which the solid state adopts a chiral helical-like conformation resembling “Siamese-Twin porphyrins”. We estimated that the energy barrier for the racemization process of the enantiomeric conformers (P/M), detected in the crystalline packing, was higher than 20 kcal mol-1.

Mechanically Interlocked Molecules
Mechanically interlocked molecules such as rotaxanes or catenanes present unique three dimensional cavities for anion recognition that resemble the preorganized pocket of anion binding proteins in Nature. The use of anions as templates in the preparation of interlocked structures has been widely described and is known to generate topologically unique cavities for anion recognition. In 2012, we reported the use of polyatomic anions for the quantitative assembly of pseudorotaxane-like architectures without involving ion-pairing in the linear component. Our approach exploited the exceptional recognition properties exhibited by a neutral interwoven self-assembled receptor towards ion-pairs. Based on this previous findings, at the beginning of this year, we reported our investigations on the synthesis of a [2]rotaxane based on a bis(calix[4]pyrrole) cyclic component and a 3,5-bis-amidepyridyl-N-oxide derivative axle. We isolated the [2]rotaxane in a significant 50 per cent yield through an optimized “in situ” capping strategy using the copper(I)-catalyzed azide–alkyne cycloaddition reaction.
The synthetic precursor of the [2]rotaxane, featuring [2]pseudorotaxane topology, could be quantitatively assembled in solution in the presence of one equivalent of tetrabutylammonium chloride or cyanate salts producing a four-particle aggregate. However, we observed that the addition of the salt was deleterious not only for the isolation of the [2]rotaxane in its pure form but, more important, for the optimal performance of the copper catalyst. We probed the interaction of the prepared [2]rotaxane with tetraalkylammonium salts of chloride, nitrate and cyanate anions by means of 1H NMR titrations and ITC experiments. We show that in chloroform solution the [2]rotaxane functions as an efficient heteroditopic receptor for the salts forming thermodynamically and kinetically highly stable ion-paired complexes with 1:1 stoichiometry. At millimolar concentration and using 1H NMR spectroscopy we observed that the addition of more than 1 equiv. of the salt induced the gradual disassembly of the 1:1 complex of the [2]rotaxane and the concomitant formation of higher stoichiometry aggregates i.e. 2:1 complexes. The reported findings augur well for the future application of water soluble analogues of the obtained [2]rotaxane as effective anion receptors in aqueous media.

Photoresponsive Dimeric Capsules
We previously reported our studies on the light-controlled assembly-disassembly of supramolecular capsules involving the synthesis of three different tetraurea calix[4]arenes equipped with four light-responsive terminal azobenzene groups. The thermally equilibrated tetraurea calix[4]arenes with all the azo groups in trans-form (all-trans isomers) dimerize quantitatively in CD2Cl2 in the presence of tetramethylphosphonium cation (Me4P+) affording a capsular aggregate. The included cation was detected by 1H and 31P NMR spectroscopy. The light induced trans-to-cis isomerization of the azo-groups produced a plethora of isomeric cis-enriched calix[4]arene dimeric capsules and triggered the partial release of the Me4P+ cation to the bulk solution. Thermal equilibration in the dark restored the system to the initial equilibrium state. We also demonstrated that we were able to control the amount of released cargo (Me4P+) after isomerization by tuning the extent of substitution of the terminal phenyl in the azobenzene groups. We were able to achieve high level of release (up to 70%) of the encapsulated guest. It is worthy to note that the switching between the thermal equilibrium state and the photostationary state proceeded with no detectable photodegradation, even when it is repeated multiple times.
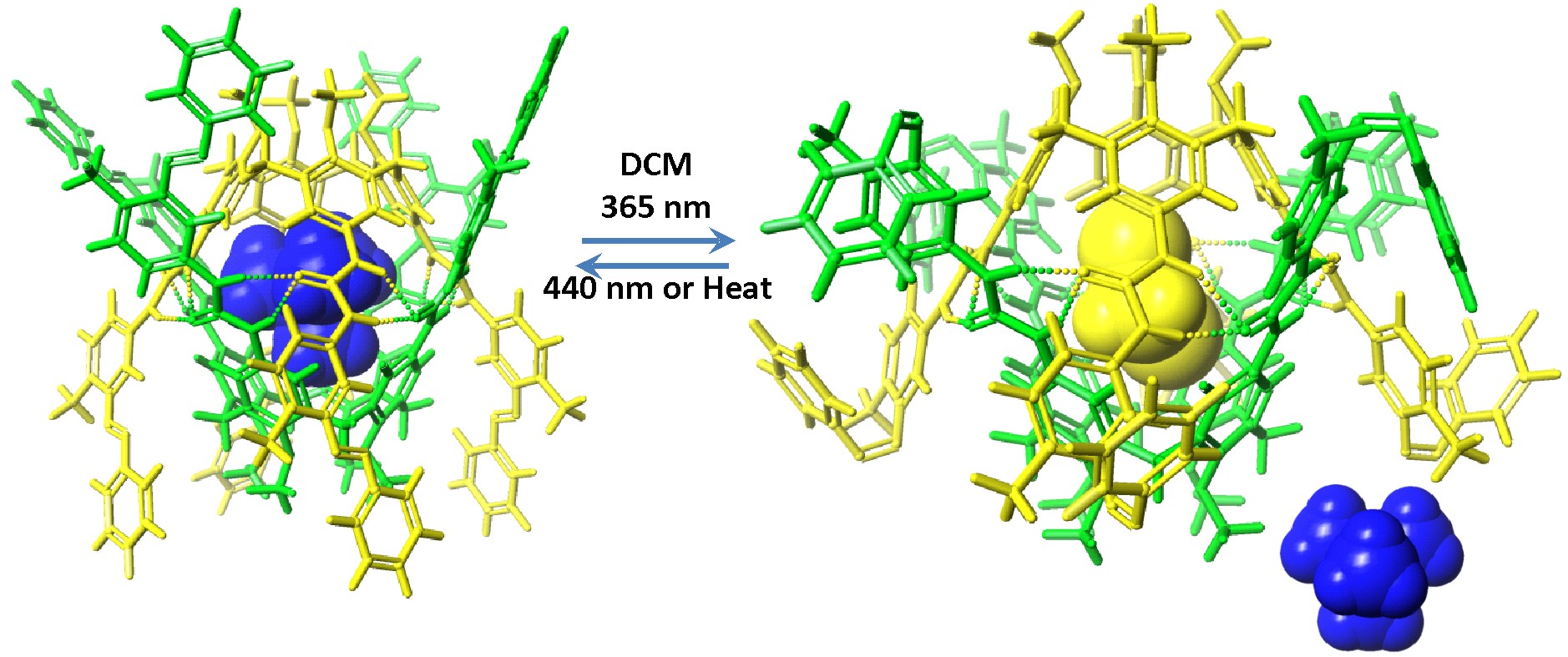
We have also described an heterodimeric capsule assembled by the social self-sorting of an all-trans calix[4]arene equipped with four terminal azobenzene units, and a calix[4]pyrrole by encapsulating one molecule of trimethylphosphine oxide or trimethylamine oxide.The trans-to-cis photoisomerisation of the azobenzene groups in all-trans-heterocapsule induced the formation of cis-enriched capsular and non-capsular assemblies that were in equilibria. The process was fully reversible by thermal treatment of the samples. Addition of bis-(N-oxide-N,N,N’,N’-tetramethylamino)hexane to the assembled heterocapsule produced the assembly of a new capsular system in the media, the homocapsule of the calixarene including the added guest. We also showed preliminary results obtained using light-irradiation for controlling the selective assembly of a capsular aggregate in complex systems.

Organic Framework based on calix[4]pyrroles
The preparation of isomeric metal–organic frameworks (MOFs) in which the network topology is controlled by the different covalent connectivity of the organic ligand is an important step forward in the design of new functional materials. In this context, macrocyclic organic ligands able to accommodate suitable guests in their own polar internal cavities are appealing candidates to act as multidentate linkers, which could potentially self-assemble into hierarchical porous structures. Taking this into account, during this year we have reported the first successful attempt to incorporate a tetraaryl-extended calix[4]pyrrole derivative into a transition metal–organic framework by simply incorporating four terminal carboxylic functional groups at the upper rim of the macrocyclic scaffold. Remarkably, the structures of the metal organic framework and its transition-metal carboxylate clusters (secondary building units, SBU) are governed by the position of the carboxylic substituent in the functionalized meso-aryl units of the linker. Only the tetra-alpha-meso-arylextended tetracarboxylic calix[4]pyrrole isomer L1, substituted in a single meta-position of their aryl rings, yields a two-dimensional MOF architecture assembled through complex Cu(II)-carboxylate clusters (SBU). The clusters have higher nuclearity than the Cu(II)2-(O2CR)4 paddle wheels produced as SBUs during the assembly of the para-substituted counterpart L2. Remarkably, the length of the dialkylformamide used as solvent (DMF or DEF) in the synthesis of the Cu(II)-organic materials derived from L2 played a key role in the structure of the final solid material. The packing of discrete metal-mediated capsular dimers of L2 switched to that of one-dimensional linear coordination polymers when the solvent’s alkyl chains were increased by a methylene unit. Finally, ligand L3, which featured a longer alkyl spacer between the para-substituted calix[4]pyrrole core and the terminal carboxylic groups than L2, self-assembled, exclusively, into discrete capsular coordination dimers also mediated by Cu(II) paddle wheel units.
The obtained results highlight the strong interplay that exists between molecular recognition, supramolecular isomerization, and solid-state structure. Conformational control of the linker substituents through binding to solvent molecules or other guests prior to self-assembly can give rise to the expected and/or unexpected solid-state architectures that may be further developed for sensing and/or catalysis applications.

Calix[4]pyrrole for sensing purposes
During these years we also focused on the application of some of the receptors developed by the group (i.e. calix[4]pyrroles and resorcinarenes) for the sensing of clinically relevant neutral and charged molecules. We can divided our studies in three different projects: (a) tetraphosphonate calix[4]pyrrole cavitands as multitopic receptors for ion-pairs and biologically relevant guests, (b) “two-wall” aryl-extended calix[4]pyrroles for the development of chloride-selective electrodes and (c) quinoxaline walled deep resrocin[4]arene cavitands self-assembled on Au-nanoparticles for benzene sensing using resistometric devices.
The first project deals with the synthesis, structural characterization, and binding studies of two disateromeric calix[4]pyrrole-resorcin[4]arene hybrids equipped with four phosphonate groups at the upper rim. The isolated tetra-phosphonate receptors displayed either three (iooo) or four (oooo) of their P=O groups oriented away from their deep and functionalized cavity. The tetraphosphonate calix[4]pyrrole cavitands exhibited superior binding properties than the previously reported bis-phosphonate counterparts in the complexation of phosphonium and ammonium ion-pairs. We demonstrated that the iooo receptor acted as a multitopic ion-pair receptor by switching the geometry of its 1:1 ion-paired complexes from a receptor-separated to close-contact mode depending on the nature of the organic cation of the chloride salt (i.e.. quaternary or primary alkylammonium).
Regarding the first project we have described an aryl-substituted calix[4]pyrrole with a monophosphonate bridge, that displays remarkable affinity for creatinine and the creatininium cation. The receptor works by including the guest in its deep and polar aromatic cavity and establishing directional interactions in three dimensions. In collaboration with the URV, we were able to incorporate into a suitable polymeric membrane this molecule to act as an ionophore. A highly sensitive and selective potentiometric sensor suitable for the determination of creatinine levels in biological fluids, such as urine or plasma, in an accurate, fast, simple, and cost-effective way has thus been developed.

In the second project of this section we used “two-wall” aryl-extended calix[4]pyrroles to prepare solid-contact ion-selective electrodes (SC-ISEs). Several calix[4]pyrrole receptors decorated with meso-phenyl substituents with different electronic properties (i.e. electron-rich, electron-poor or electron-neutral) were incorporated as ionophores in SC-ISEs membranes. The ISEs also incorporated multiwall carbon nanotubes (MWCNTs) as efficient transducers. The “two-wall” calix[4]pyrrole bearing a p-nitro electron-withdrawing group on each of the meso-phenyl rings, afforded sensors that displayed anti-Hofmeister behaviour against lipophilic salicylate and nitrate anions. Moreover, we demonstrated that this receptor was an excellent ionophore towards chloride anions. The concerted use of hydrogen-bond and anion-π interactions in the formation of the complex provided an ISE with excellent selectivity for chloride against common and highly lipophilic anions. The developed sensors were used to measure chloride concentration in real and artificial biological samples of blood, serum, sweat and urine, obtaining excellent recovery.
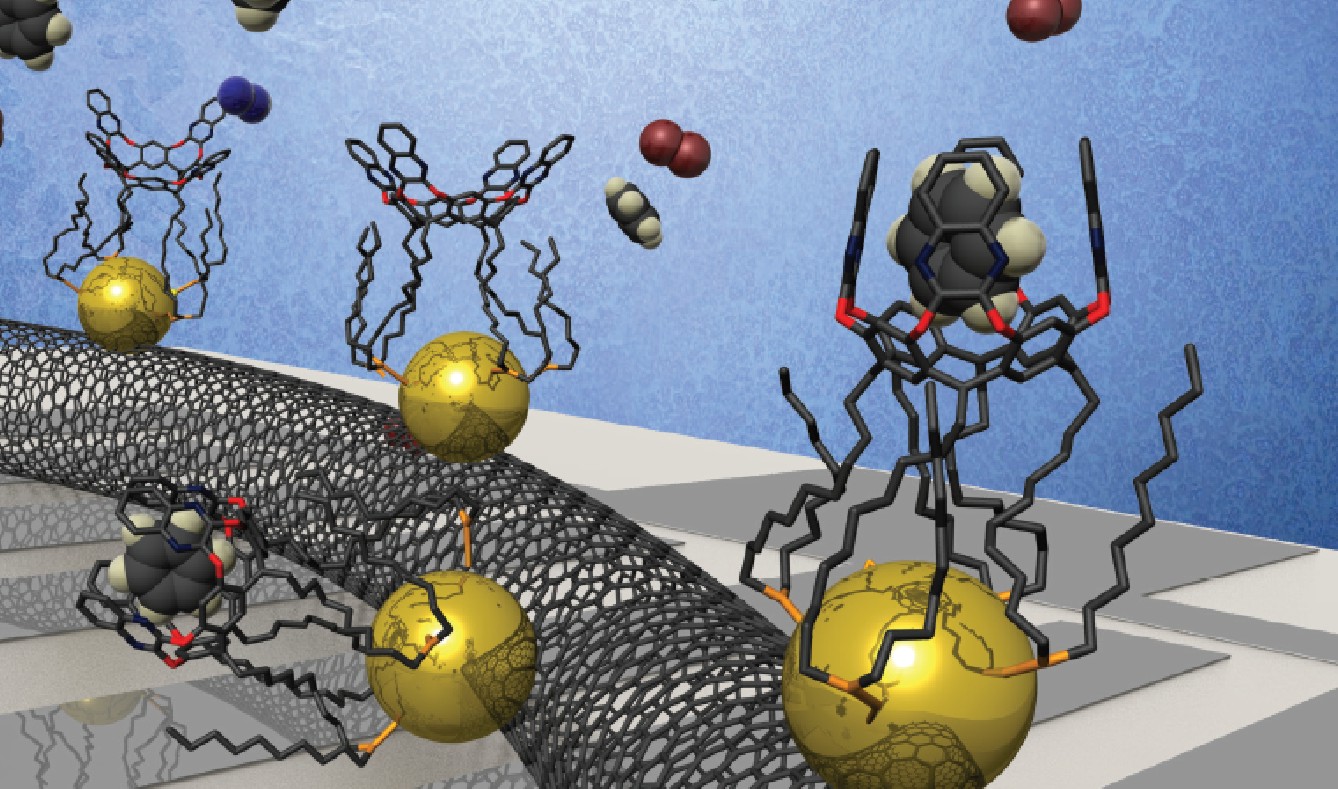
Finally, in the last project centred on sensing devices we developed, in collaboration with the group of Prof. E. Llobet from the URV, a simple experimental procedure for preparing a resistive gas sensor device. The sensor contained gold nanoparticles (AuNPs) deposited on MWCNTs. In turn, the AuNPs were decorated with a quinoxaline-walled thioether-legged resorcinarene cavitand (cav). The cav-AuNP-MWCNT resistive gas sensor showed outstanding performance toward traces of benzene vapours. The detection of 2.5 ppb of benzene in dry air was demonstrated with a limit of detection (LOD) near 600 ppt. Moreover, the sensor response toward toluene and o-xylene was significantly lower, confirming the strong response for benzene.